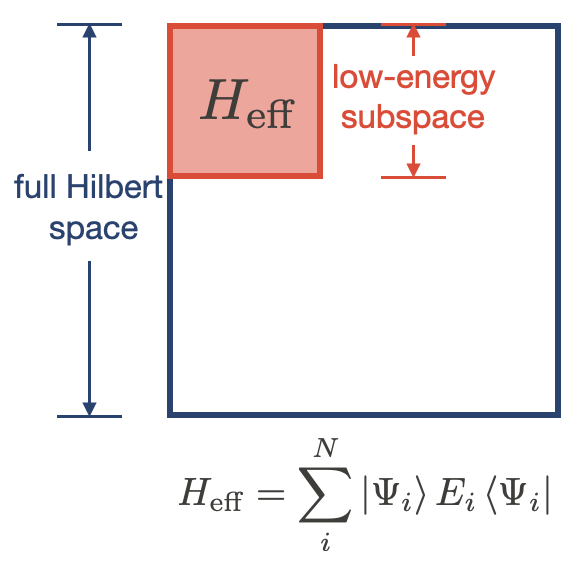
I am currently working on developing downfolding methods, to derive effective models for many-electron systems from highly accurated first principles.
Reach me at yueqingchang AT gmail DOT com
Research projects
Effective spin-orbit models using correlated first-principles wave functions
Phys. Rev. Research 2, 013195 (2020)
Yueqing Chang and Lucas K. Wagner
Diffusion Monte Carlo using continuous real-space wave functions is one of the most accurate scalable manybody methods for solid-state systems. However, to date, spin-orbit interactions have not been incorporated into large-scale calculations at a first-principles level, only having been applied to small systems. In this technique, we use explicitly correlated first-principles diffusion Monte Carlo calculations to derive an effective spin-orbit model Hamiltonian. The simplified model Hamiltonian is then solved to obtain the energetics of the system. To demonstrate this method, benchmark studies are performed in main-group atoms and monolayer tungsten disulfide, where high accuracy is obtained.
Nanoscale studies of electric field effects on monolayer 1T’-WTe2
npj Quantum Mater. 7, 29 (2022)
Yulia Maximenko, Yueqing Chang, Guannan Chen, Mark R. Hirsbrunner, Waclaw Swiech, Taylor L. Hughes, Lucas K. Wagner, Vidya Madhavan
Monolayer 1T’-WTe2 is a quantum spin Hall insulator with a well-defined bulk gap and helical edge states. It has been predicted to undergo a topological phase transition upon breaking the inversion symmetry with gating [1]. Recent scanning tunneling microscopy (STM) experiments by Maximenko et. al. have found a surprising linear dependence of the gap on gating voltage. We use first principles to study the electronic structure of monolayer 1T’-WTe2 with the effect of gating, using a tight-binding model derived from the Wannierised Kohn-Sham orbitals computed from density functional theory. We find that the top and bottom surfaces of the monolayer 1T’-WTe2 exhibit opposite spin-momentum locking properties. This effect causes the STM measured gap to show a linear response upon gating.
References:
[1] X. Qian, J. Liu, L. Fu, J. Li, Science 346.6215 (2014).
Minimal effective models from first principles for hydrogen chains in the strongly correlated regime
One powerful application of first-principles calculations is to provide effective low-energy descriptions of materials (downfolding). This downfolding is commonly done by assuming a given model, then estimating the model parameters using first-principles results. Examples include computing J using different spin orders, the cRPA-based approaches in which a particular subspace and interaction formulation are chosen, and modifying DFT band structures with interactions. While they can be successful, these approaches are not systematically improvable.
The density matrix downfolding (DMD) approach was proposed [1,2] to resolve this issue. DMD treats the non-interacting and interacting terms all on the same footing. It allows one to systematically improve the downfolded Hamiltonians using more accurate approximations to the Hilbert space, and better parameterizations of the Hamiltonians. In this study, we apply DMD to hydrogen chains in the strongly correlated regime. Specifically, we show that DMD naturally selects the only relevant parameters in the model for the chosen Hilbert space, with all the parameters renormalized from the bare long-range values. For the hydrogen chains, we show that the minimal effective model on a low-energy Hilbert space spanned by the first few spin-like excitations is a Heisenberg model. The minimal model becomes a single-band Hubbard model when extending the Hilbert space to include the charge excitations.
References:
[1] H. J. Changlani, H. Zheng, and L. K. Wagner, J. Chem. Phys. 143, 102814 (2015).
[2] H. Zheng et. al., Front. Phys. 6, 43 (2018).
Electron-phonon coupled effective Hamiltonians derived using correlated many-body wave function methods
In some low dimensional meterials, interaction with the collective motion of the atoms leads to a type of ground state called charge density wave, where certain distortions of the lattice are allowed upon lowering temperature. Bulk 1T-TiSe2 shows a 2X2 CDW transition at around 232 K, and a 2X2X2 transition at around 205 K. Several scenarios have been proposed to explain the origin of the CDW formation: the band Jahn-Teller effect, strong electron-phonon coupling, exciton insulator formed by exciton condensate, and a collaboration between exciton condensation and electron-phonon coupling. To our knowledge, no first-principles study that is focused on the effect of electron-phonon coupling in 1T-TiSe2 has ever been reported. Besides, the cooperation of exciton formation and electron-phonon coupling has not been covered on the level of highly accurate first-principles studies. How the energy drop is contributed by electron-phonon coupling and exciton condensation remains quantitatively undetermined.
With density-matrix downfolding method based on quantum Monte Carlo calculations, we can derive a model Hamiltonian for 1T-TiSe2, both normal and CDW phases. This enables us to disentangle the multiple mechanisms underlying the formation of CDW, by attributing the energy drop to the corresponding terms in the Hamiltonian.